Diagnosis and Treatment
Diagnosis
Although HD involves many complex alterations in the brain, a simple blood test can confirm if a mutant copy of the disease gene is present; extensive medical and physical tests are combined with family and medical histories to complete the diagnosis.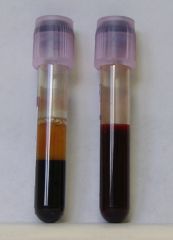
The test can be carried out if there is a family history of the disease, if a person is suffering from apparent symptoms of the disease, or alternatively if a person is considering having children and is worried about passing a faulty copy of the Htt gene onto their offspring.
As the gene position is already known to be IT-15 on Chromosome 4, this makes genetic testing simpler as researchers know where the gene will be found. A person is considered as having HD if the number of CAG or PolyQ repeats exceeds 38, with a wild-type person having up to 34. There is therefore a grey-area in between, which sometimes makes it harder to give a complete diagnosis. However with a full family history, and often a blood sample being taken from a close relative, it is possible to give a reliable diagnosis in the majority of cases.
Presymptomatic testing can even be done on a foetus in the womb if it is seen necessary.
Photograph of two blood samples. Image courtesy of commons.wikimedia.org/wiki/File:Blut-EDTA.jpg under the GNU Free Documentation License.
Brain scans can also confirm a diagnosis, as the disease is usually associated with a decrease in the size of the caudate nucleus and putamen, as well as enlargement of ventricles. The diagram below highlights some of the key areas affected.
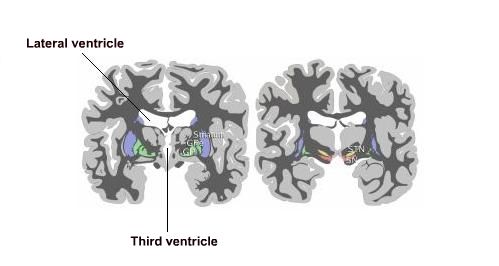
Coronal sections of the basal ganglia. Image adapted courtesy of commons.wikimedia.org/wiki/File:Basal-ganglia-coronal-sections.png under the GNU Free Documentation License.
However, a decrease in size of the caudate nucleus and putamen, or an increase in ventricle size does not necessarily equate to having the disease; just as having the disease does not always lead to these results. This is why genetic tests are needed for a complete diagnosis.
Prevalence
HD is the most commonly inherited of PolyQ-associated diseases, occurring in 1 in 10,000 people. Other associated diseases include spinobulbar muscular atrophy (SBMA) and spinocerebellar ataxia types 1, 2, 3, 6, 7 and 17.
Currently there is no cure for Huntington's disease. However medication, therapy and diet can help to treat some of the symptoms.
Treatment
Future Methods
New methods of treating the disease is a constant source of research throughout the world, below are some therapeutic methods that could prove effective in the future.
Removal of mutant Htt
This can be done by RNA interference methods.
This would in fact be the best cure for the disease, however due to the lack of safe gene delivery methods, the success of this method is still a very long way off.
Blocking the interactions of the mutant Htt with transcription factors
This would therefore prevent the toxic affect of the mutant Htt in the nucleus.
This is currently hard to achieve and therefore a long way off still.
Genetic and chemical agonists and antagonists
Aimed at specific transcription factors such as p53 (which regulates the cell cycle), NF-kB (a protein complex that acts as a transcription factor, important for learning and memory) and CBP (CREB binding protein; CREB acts as a transcription factor); altering their regulatory pathways.
Photograph of a lab mouse. Image courtesy of commons.wikimedia.org/wiki/File:Lab_mouse_mg_3263.jpg under the Creative Commons "Attribution ShareAlike 2.0 France" license.
Histone acetylation
Increasing the amount of histone acetylation by targeting enzymes responsible for histone deacetylation, using small molecular inhibitors.
Selective inhibitors will need to be developed in order to prove most effective, but developing these specific CNS inhibitors has proved to be slow so far. One example being closely studied is resveratrol, which has been shown to exhibit protective effects on neurons.
Chromatin and DNA binding modifications
Small DNA intercalators, for example Mithromycin-A could block the mutant Htt from binding to the DNA, preventing the repression of transcription.
Targeting REST
REST is a transcriptional repressor that is normally sequestered by the Htt protein, allowing BDNF to remain at the physiological level; mutant Htt releases this REST and prevents transcription of BDNF, therefore decreasing the available levels of BDNF.
Targeting REST could therefore prevent the depressed levels of BDNF as seen in HD, and restore the normal levels.
Cell-based therapy
Primary foetal cells transplanted into the striatum in rats and mice led to improvement in some behavioural-associated symptoms, due to the formation of functional connections. Other potential sources of stem cells include adult CNS stem cells, non-neuronal stem cells such as from the umbilical cord or bone marrow, plus embryonic germ cells
- Foetal neural stem cells- neural potential has been seen to decline, but they are able to respond to a variety of signals from the CNS. Methods need to be developed to push forward the striatal phenotype before transplantation.
- Adult neural stem cells- can develop into mature neurons, oligodendrocytes and astrocytes in vitro; more work needs to be done in order to produce the same effect in vivo.
- Embryonic stem cells- isolated from the inner cell mass of a blastocyst; specific neural progenitor cells have been derived in culture. So far there is no evidence of differentiation into striatal cells.
- Embryonic germ cells- from primordial germ cells of the gonadal ridge in early-stage embryos. Appear to contain neural progenitors, but not enough work has been done with them so far.
- Non-neural stem cells- bone marrow stem cells have been reported as having the capacity of differentiating into neurons in rodents. The full capacity of these cells is not yet understood.
With such a diverse range of pathways implicated in the disease and available for drug targeting, it will be a long time before a suitable therapeutic method is close to being finalised.
Image of stem cell division and differentiation. Image courtesy of commons.wikimedia.org/wiki/File:Stem_cell_division_and_differentiation.svg.
Current methods
At the minute, with no effective cure for HD, treatment methods aim to alleviate some of the known symptoms of the disease.
Medication
- Antipsychotics, including chlorpromazine and haloperidol
- Antidepressants, including fluoxetine, sertraline and nortriptyline
- Tranquilizers, including benzodiazepines and beta-blockers
- Mood-stabilizers, including lithium and valproate
- Botulinum toxin (to alleviate dystonia and jaw clenching)
Photograph of medication. Image adapted courtesy of commons.wikimedia.org/wiki/File:Gelcaps.jpg under the Creative Commons Attribution ShareAlike 2.5 license.
Diet
A high calorie diet is required in order to maintain body weight and a larger volume of fluids than normal to prevent dehydration.
Loss of movement coordination makes eating and drinking difficult in HD sufferers.
Physical and social activity
These simply maintain the patient's physical and mental well-being; patients are encouraged to keep up both activities until it is no longer possible for them to do so.
A lot of patients are looked after by professional carers.