Causes
This section aims to look at the main cause of Huntington's disease at a molecular level.
The Huntingtin protein
Huntingtin (Htt) is a protein ubiquitously expressed throughout the brain and body. It is a large protein and its normal role is not very well understood. It contains regions of Poly-glutamine (PolyQ) repeats in its N-terminus; the normal copy of the protein carries between 11 and 34 of these repeats, however the mutant copy carries over 37 and is inevitably linked to Huntington's disease. Juvenile HD sufferers have around 60 of these repeats.
The Htt protein is coded for by the Huntingtin gene, of which the mutant copy has multiple CAG repeats.
The mutant gene IT-15 is found on chromosome 4.

Picture adapted from Linder and Deschenes (2007).
The expansion of this PolyQ region leads to aggregation of the protein and neurotoxicity. The mutant protein particularly accumulates in the nucleus and processes of the neuron; accumulation in the nucleus therefore leads to changes in gene expression which can prevent the neuron from functioning normally. There are three suggested mechanisms of gene disruption:
1) Disruption of the nuclear matrix
2) Disruption of certain gene transcription mechanisms
3) Sequestering of certain proteins into a complex
Htt is also said to be associated with transport proteins such as microtubules and intracellular vesicles due to impaired transport mechanisms in sufferers.
Aggregation of the Htt protein eventually results in neurodegeneration in certain areas, mainly the striatum and deep areas of the cortex; as discussed in the 'overview' section these areas play important roles in the control of movement.
Later, neurodegeneration also occurs in the hippocampus, hypothalamus- specifically the lateral tuberal nucleus, cerebellum, amygdala and thalamic nuclei.
The table below shows some of the key roles played by the above mentioned areas, and gives us an idea as to the symptoms of HD.
|
Area |
Normal function |
|
Striatum |
Part of an inhibitory pathway to the globus pallidus; releases inhibition of the thalamus, leading to the initiation of movement |
|
Hippocampus |
Mainly memory functions |
|
Hypothalamus- lateral tuberal nucleus |
Thirst and hunger functions |
|
Cerebellum |
Movement control; generates the overall background movement, timing and force of the movement |
|
Amygdala |
Implicated in aggression, emotion, fear and regulation of the hypothalamic-pituitary-adrenal (HPA) axis |
|
Thalamic nuclei |
A variety of roles, including emotion, learning, memory, cognition and alertness |
The worst affected neurons are the projection neurons, such as the medium-sized spiny neurons of the striatum. These are GABA-ergic neurons that innervate the substantia nigra and the globus pallidus.
Cytoplasmic Htt aggregation can also cause cytotoxixity.
The PolyQ repeats are as a result of an expansion of the CAG repeat in the coding region of the gene. There is a positive correlation between the number of these repeats and the age of onset of the disease, and this has also been suggested to correlate with the rate of progression. However the duration of the disease itself is said to be independent of this matter.
Models of human disease
Mice are important biological systems used to study a lot of human diseases; being mammals there is a large amount of homology between signaling pathways and protein events with humans. They allow the early pathological changes of the disease to be studied, as can not otherwise be done in humans.
Mice with mutant Htt are said to have a 'gain of toxicity' mutation. This mutated protein becomes misfolded and aggregates, binding irregularly to other proteins or forming a separate complex that then interferes with cellular functions. It has been suggested that the PolyQ expansion could allow the protein to mimic transcription factors and directly influence gene transcription.
It was also discovered that expansion of the PolyQ segment can lead to apoptosis; this is because it affects cell survival signaling pathways, prevents the removal of toxic segments and affects gene expression if it is present in the nucleus.
Mutant Htt also affects the expression of the brain derived neurotrophic factor (BDNF), which promotes survival of striatal neurons, as well as activating caspase signaling pathways, such as caspase-8, which lead to call death.
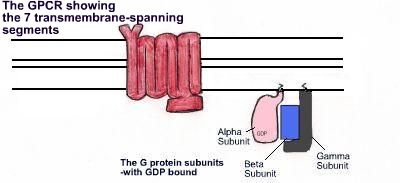
It has been observed in transgenic mice that there is a decrease in the level of certain neurotransmitter receptor mRNA at around four weeks of age, specifically D1 and D2 dopamine receptors (GPCRs), A2 receptors for adenosine, group I and group II metabotropic glutamate receptors, and ionotropic kainate and AMPA glutamate receptors. These receptors will all play important roles in the basal ganglia, and throughout the rest of the brain. The fact that these mRNA levels were low before symptoms of the disease occurred in transgenic mice, suggests that this is a cause rather than a secondary consequence of the disease.
One observed feature in mutants is a decrease in histone acetylation, which causes transcriptional silencing and could therefore account for the transcriptional depression in HD sufferers.
Inheritance
As the disease is inherited in an autosomal dominant manner, a child with an affected parent has a 50/50 chance of getting the disease themselves (as demonstrated below).
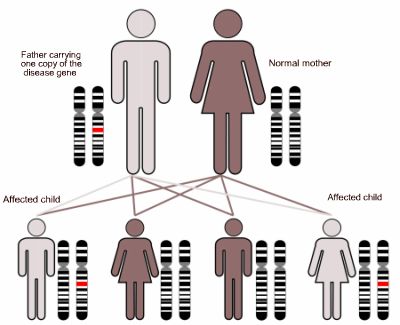
Diagram demonstrating an autosomal dominant method of inheritance. Image adapted from commons.wikimedia.org/wiki/File:Autodominant_01.png under the terms of Creative Commons Attribution ShareAlike 3.0 license.
Onset of the disease normally occurs between the ages of 30 and 50. However Juvenile Huntington's disease can occur in people below the age of 20, but this only occurs in around 5% of sufferers.
Research shows that there is no direct relationship between the level of Htt in the striatal neurons and a person's vulnerability to the disease, meaning that other internal and external factors are in fact involved.